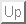

1. INTRODUCTION
Protein motions and functional processes in proteins
occur on a wide range of time scales. The fastest atomic motions
take place on a femtosecond time scale.
Fast biochemical reactions like the primary steps in
photosynthesis last few picoseconds. Most biochemical reactions like
enzymatic processes take much longer --- microseconds or even few
milliseconds. They are often accompanied by larger structural
rearrangements in the protein, called conformational transitions[1],
which are characterized by transition times of nanoseconds or much longer.
A prominent example for an extremely slow conformational transition, with
a transition time of many years, is the
one which is believed to be responsible for the pathogenic effect of
prions[2]. Often, conformational transitions constitute functional
important motions, as for the gating of channel proteins or in protein folding.
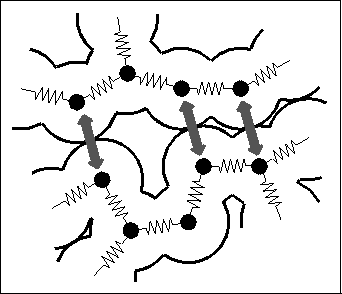
Figure 1: Molecular dynamics (MD) simulations serve to describe atomic
motions in proteins[3]
and other biological macromolecules. In this
semiclassical approach, a protein is modeled as a set of point
masses (atoms). The interatomic forces are described by a semiempirical
energy function, and the molecular dynamics is determined
by numerical integration of the Newtonian equations of motion.
Typical forces included in the energy function are chemical binding
forces (`springs') as well as noncovalent interactions like Pauli repulsion
(grey arrows), Van der Waals- and Coulomb-forces
(see, e.g., Ref.[4]). A good review on the MD-method is
Ref.[5].
Most of these processes are not understood on an atomic level. Therefore, adequate descriptions of their molecular dynamics (MD) are required in the attempt to derive function from structure. Due to the structural complexity of proteins and a corresponding lack of well-founded coarse-grained effective models for the dynamics, the method of MD-simulation[6,3] currently is the only approach, to which some reliability can be assigned. That method (see Figure 1) conceives a macromolecule as a classical many-body system of `atoms' and describes the quantum-mechanical forces like the chemical binding forces, which are caused by the electronic degrees of freedom, by a semi-empirical force field. Accordingly, the molecular dynamics is simulated by integration of the Newtonian equations of motion.
However, the enormous computational task associated with MD-simulation of biological macromolecules entails an upper limit to the time scale of dynamical processes accessible by this method: At present, only few nanoseconds of protein dynamics can be simulated. Extrapolating the past increase of available computer power, one is forced to the conclusion, that we will not be able to enter the biochemically most relevant microsecond time scale within the next ten years, if we cannot manage to drastically reduce the necessary amount of computations. Also, protein folding simulations, which might contribute to solve the challenging `folding problem', will be out of range for quite a while.
Given this situation, it is worthwhile to look for new concepts in order to simplify the description of protein dynamics. Clearly, the identification and elimination of irrelevant degrees of freedom would be particularly helpful. In this report, we will describe the stage, on which, as far as we believe, the necessary simplifications will take place. Transitions between conformational substates, which are ubiquitous in proteins[7] play a central role here, as they mediate between detailed atomic motions, as described by MD-simulations on the one hand and, on the other, the biochemical reactions we want to study. For this reason, those degrees of freedom, which describe the conformational dynamics of proteins --- conformational coordinates --- are of special interest. Unfortunately, conformational transitions have rarely been observed in MD-studies, since the latter are usually too short. Hence, it is not clear how to identify suitable conformational coordinates.
Having introduced the concepts of `configurational space' and `projected
configurational space densities', we will suggest two methods
to construct conformational coordinates. The first one describes
conformational transitions on time scales above nanoseconds and is
based on a neural clustering algorithm. The second method aims at a
description of conformational motion within configurational
substates on a faster time scale.
2. A SIMPLIFIED PROTEIN MODEL
In order to illustrate these methods, we use a simplified protein
model[] (Fig. 2)
with a reduced number of degrees of freedom.
It has been designed to fulfill two contradicting requirements:
(i) the model should be structurally and dynamically similar
to real proteins; in particular, it should exhibit conformational
transitions; (ii) the model should have as few degrees of freedom as possible
in order to allow extended MD-simulations. In that sense, it is a
minimal model.
Figure 2: Simplified protein model used in the analysis described in the text.
The model consists of 100 point masses (not drawn), which are covalently
linked by chemical bonds. Like the residues in a real protein, the 100 model
residues differ in their interaction properties. A stable tertiary
structure of the model system (shown as a `ribbon-plot') has been
obtained from a simulated folding process of  ns duration.
ns duration.
Accordingly, a primary structure consisting of 100 amino acids was defined, where each of the residues is modeled as a single van der Waals sphere. Covalent and non-covalent interactions were added to account for the dominant forces known to stabilize the tertiary structure of proteins, such as chemical binding forces or hydrophilic/hydrophobic interactions. Starting from a `denatured' configuration, a folding process of 5 ns length has been simulated, which yielded the stable and equilibrated tertiary structure sketched in Fig. 2. As shown in Ref.[8], the model meets the above two criteria.
MD-simulations of a total of 232 ns have been carried out on that
protein model. We will use results of these simulations to illustrate concepts
and methods to study the conformational dynamics of proteins.
3. CONFIGURATIONAL SPACE AND CONFORMATIONAL SUBSTATES
We start with the concept of configurational space, which is closely
related to the concept of phase space. Fig. 3
illustrates the basic notions.

Figure: Protein structure and dynamics can be analyzed in configurational
space. (a): A given protein structure described
by N atomic positions
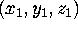 ,
,
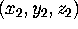 , ... ,
, ... ,
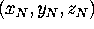 (left side) is mapped into configurational space
by merging the N vectors into one single vector
(left side) is mapped into configurational space
by merging the N vectors into one single vector
 with dimension
3N
(right side). (b): Accordingly, the concerted motion of all atoms
in a protein can be described by one single high-dimensional trajectory in
configurational space. (c): Snapshots of protein dynamics, e.g. derived from
an MD-simulation, are mapped into a `cloud' of configurations, where
each point in configurational space resembles the particular geometry
of the protein at one instance in time. If a sufficiently large sample
of configurations is available, one can estimate the configuration space
density, which is generated by the molecular dynamics. By projecting
this high-dimensional density on a suitable low-dimensional subspace
one can gain insights into the overall structure of this density
(contour lines).
with dimension
3N
(right side). (b): Accordingly, the concerted motion of all atoms
in a protein can be described by one single high-dimensional trajectory in
configurational space. (c): Snapshots of protein dynamics, e.g. derived from
an MD-simulation, are mapped into a `cloud' of configurations, where
each point in configurational space resembles the particular geometry
of the protein at one instance in time. If a sufficiently large sample
of configurations is available, one can estimate the configuration space
density, which is generated by the molecular dynamics. By projecting
this high-dimensional density on a suitable low-dimensional subspace
one can gain insights into the overall structure of this density
(contour lines).
At an instant of time, the three-dimensional structure of a protein
consisting of N atoms is defined by
N atomic positions, i.e.,
N three-dimensional vectors (Fig. 3 (a), left side).
Alternatively, the structure can be
represented by one single 3N-dimensional vector, which is obtained
by combining the N atomic positions. In this picture, one
single point in high-dimensional configurational space defines
all atomic positions of the protein (Fig. 3 (a),
right side).
For typical proteins, configurations space has some 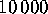 dimensions;
for the simplified protein model described in the preceding section,
configurational space is of dimension 300.
dimensions;
for the simplified protein model described in the preceding section,
configurational space is of dimension 300.
The conceptual
advantage of looking at a protein in configurational space becomes
clear if we switch from protein structure to dynamics. Then,
the concerted motion of all atoms of the protein
(Fig. 3 (b), left side) is represented
by one single trajectory in configurational space
(Fig. 3 (b), right side). An MD-simulation typically
provides a set of `snapshots' of that atomic motion
(Fig. 3 (c), left side), which appears
in configurational space as a `cloud' of configuration vectors
(Fig. 3 (c), right side). In the limit of
large numbers of configurations a `configuration space density' 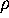 ,
as determined by the protein dynamics, can be derived.
,
as determined by the protein dynamics, can be derived.
Now consider a few (m) degrees of freedom,
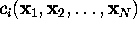 (
(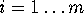 ),
called `conformational coordinates'. For a given protein, these
degrees of freedom shall be chosen such, that they reflect
the conformational dynamics of that protein. Then, a coarse-grained
effective description of that (microcanonical) system with
Hamiltonian
),
called `conformational coordinates'. For a given protein, these
degrees of freedom shall be chosen such, that they reflect
the conformational dynamics of that protein. Then, a coarse-grained
effective description of that (microcanonical) system with
Hamiltonian 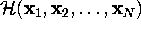 can be achieved by explicitly considering the dynamics of only
the
can be achieved by explicitly considering the dynamics of only
the 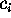 . If the remaining
3N-m degrees of freedom are regarded
as a heat bath, the resulting system of reduced dimension
belongs to a canonical ensemble.
. If the remaining
3N-m degrees of freedom are regarded
as a heat bath, the resulting system of reduced dimension
belongs to a canonical ensemble.
The free energy landscape of that sub-system, 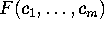 ,
determines, together with the heat bath, the dynamics of the
conformational coordinates
,
determines, together with the heat bath, the dynamics of the
conformational coordinates 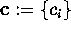 .
.
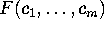 can be derived from the configurational space density
can be derived from the configurational space density
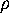 by
by
where 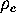 is a canonical conformational space density,
obtained by projecting
is a canonical conformational space density,
obtained by projecting  on the
m-dimensional subspace
spanned by the conformational coordinates
on the
m-dimensional subspace
spanned by the conformational coordinates 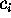 (Fig. 3 (c), right side),
(Fig. 3 (c), right side),
As an illustration, Figure 4
shows a contour-plot of the free energy landscape of our protein
model, which has been determined from
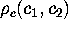 according to (1).
The inset of Fig. 4 illustrates the shape of the
free energy landscape F
by plotting its value in units of
according to (1).
The inset of Fig. 4 illustrates the shape of the
free energy landscape F
by plotting its value in units of 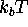 (at T=300K)
along an arbitrarily chosen `reaction coordinate'
(bold line) passing through the three minima.
We did not yet
describe how the two conformational coordinates
(at T=300K)
along an arbitrarily chosen `reaction coordinate'
(bold line) passing through the three minima.
We did not yet
describe how the two conformational coordinates 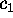 and
and
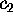 have been defined; we will do that in the following section.
have been defined; we will do that in the following section.
The energy landscape in Fig. 4 exhibits three distinct minima, denoted as `B', `C', and `D', respectively. As suggested on the basis of experimental data by Frauenfelder[9] such regions of low free energy in conformational space, separated by free energy barriers, generally define distinct conformational substates of a protein. Accordingly, we define our model to be in substate `B', `C', or `D', respectively, if its conformation lies in the corresponding region of the free energy landscape. Like the Brownian motion of a particle coupled to a heat bath, the dynamics of the system within the energy landscape shown in Fig. 4 is diffusive. Occasionally, the fluctuating forces generated by the heat bath drive the system across one of the energy barriers and induce a conformational transition, which reveals itself as a rapid change in the sterical structure of the model.
Note, that the above definition of conformational substates
differs from the approach commonly employed for
theoretical explorations of
substate hierarchies[10,11]. In these studies,
the distribution of
thermally accessible local minima of potential energy
within configurational space is studied, and
it is assumed, that these local minima or clusters thereof
can provide information
on the distribution of conformational substates. At low temperatures,
where entropic contributions are small and safely can be neglected,
the potential energy landscape definitely can
serve as a tool for the analysis of conformational substates.
However, at room temperature the suggested relation between
accessible minima of potential energy and conformational substates,
defined as minima of free energy, is questionable.
In contrast, our approach allows
the study of conformational substates at physiological temperatures,
as it refers to the free energy landscape within conformational space.
Therefore,
if applied to realistic protein models, our approach enables
comparisons of theory and experiment.
Admittedly, much larger computational effort is involved in such analysis,
because a sufficiently dense sampling of phase space by extended
simulations is required for the determination of 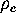 .
At present, that computational effort restricts our method to studies of
simplified protein models.
.
At present, that computational effort restricts our method to studies of
simplified protein models.
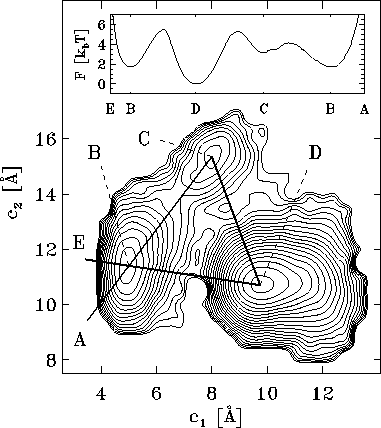
Figure 4: Free energy landscape derived from a 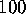 ns simulation
of a simplified protein model, shown as a contour plot.
It has been determined by projecting the configuration space
density
ns simulation
of a simplified protein model, shown as a contour plot.
It has been determined by projecting the configuration space
density 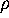 onto onto the two-dimensional subspace defined by the
conformational coordinates
onto onto the two-dimensional subspace defined by the
conformational coordinates 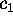 and
and
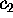 (see text).
The energy surface exhibits three minima, denoted by `B', `C', and `D',
which define three configurational substates. The inset shows
a cross section through the energy landscape along a hypothetical
reaction coordinate E-B-D-C-B-A in units of
(see text).
The energy surface exhibits three minima, denoted by `B', `C', and `D',
which define three configurational substates. The inset shows
a cross section through the energy landscape along a hypothetical
reaction coordinate E-B-D-C-B-A in units of 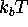 at
at
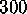 K.
K.
We now return to the question, how suitable conformational
coordinated can be determined. Clearly, the quality of a
dimension-reduced description of protein dynamics depends
on the choice of conformational coordinates.
We studied two methods[12], which work on different
levels of coarse graining. The first method, sketched in the following
section, serves to identify conformational substates on the
basis of MD-data and aims at a description of conformational
transitions. The second method, described in Section 5, can be used
to construct an effective description of the molecular dynamics
within a conformational substate.
4. CONFORMATIONAL COORDINATES FROM NEURAL CLUSTERING
We defined conformational substates as regions in
configurational space of low free energy, or, according to Eq.1,
of high configuration space density. Hence, statistical
data analysis should provide tools that can be used to find clusters
in a `cloud' of high dimensional configuration vectors. As
conformational substates are known to be hierarchically
structured[13], such a tool should be able to
reveal cluster hierarchies.
For that purpose we used a neural cluster algorithm (`minimum free energy
clustering'), which has recently been proposed[14].
We will not go into details of this algorithm here, but
show merely the results of its application to a 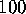 ns MD-simulations
of our simplified protein model.
ns MD-simulations
of our simplified protein model.
The algorithms takes as input a (typically large) set of configurational
vectors. In our example, a total of 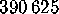 configurational vectors
were used, sampled at intervals of
configurational vectors
were used, sampled at intervals of 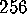 fs. Each of the six
pictures in Fig. 5 shows as a contour plot
a projection of the configurational space density defined by these
configurational vectors similar to the one shown in Fig. 4.
Here, too the three substates A, B, and C, respectively, can be
identified (Fig. 5 (a)). The algorithm then
finds minima in a `blurred' energy landscape, which is, approximately,
a convolution of the free energy
fs. Each of the six
pictures in Fig. 5 shows as a contour plot
a projection of the configurational space density defined by these
configurational vectors similar to the one shown in Fig. 4.
Here, too the three substates A, B, and C, respectively, can be
identified (Fig. 5 (a)). The algorithm then
finds minima in a `blurred' energy landscape, which is, approximately,
a convolution of the free energy 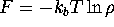 with a
multidimensional Gaussian of width
with a
multidimensional Gaussian of width 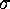 . By variation of the
length scale
. By variation of the
length scale 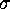 , one can conveniently analyze the hierarchical
structure of substates.
, one can conveniently analyze the hierarchical
structure of substates.
Figure: The structure of high dimensional configuration space densities
can be analyzed using hierarchical neural clustering algorithms.
The sequence of six pictures (a-f) shows how such an algorithm
can resolve structures of the density on decreasing length scales
[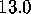 Å (a),
Å (a),
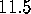 Å (b),
Å (b),
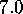 Å (c),
Å (c),
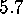 Å (d),
Å (d),
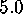 Å (e),
Å (e),
 Å (f)]:
Regions of high configuration space density, as detected by the
clustering algorithm, are marked as triangles; superimposed is
a contour plot of a suitable configuration space density projection similar
to that shown in Fig.4.
Å (f)]:
Regions of high configuration space density, as detected by the
clustering algorithm, are marked as triangles; superimposed is
a contour plot of a suitable configuration space density projection similar
to that shown in Fig.4.
The sequence of six pictures in Fig. 5 shows
the output of the clustering algorithm (triangles)
for different length scales 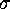 . For
. For
 as large as
as large as
 Å (Fig. 5 (a))
the three clusters cannot be resolved and are identified as one single
cluster positioned at the center of mass of the data set. In picture (b),
as
Å (Fig. 5 (a))
the three clusters cannot be resolved and are identified as one single
cluster positioned at the center of mass of the data set. In picture (b),
as  is decreased,
a bifurcation occurs, and the single minimum splits into three
distinct minima representing the three conformational states (c).
Below
is decreased,
a bifurcation occurs, and the single minimum splits into three
distinct minima representing the three conformational states (c).
Below 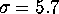 Å the conformational states exhibit further
substructures, which we will not discuss here.
Å the conformational states exhibit further
substructures, which we will not discuss here.
Now we are prepared to define the conformational coordinates 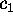 and
and
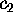 , which we used in Figs. 4 and 5
in order to visualize the high dimensional configurational space density
, which we used in Figs. 4 and 5
in order to visualize the high dimensional configurational space density
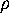 : The three free energy minima identified
with the neural clustering algorithm define a plane, onto which
: The three free energy minima identified
with the neural clustering algorithm define a plane, onto which  has been projected. Accordingly, the conformational coordinates
has been projected. Accordingly, the conformational coordinates  and
and
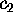 have been chosen to lie within this plane. Hence projections of
have been chosen to lie within this plane. Hence projections of
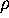 on that subspace optimally resolve the three substates. Note that it
would have not been possible to
identify number and location of the three substates
by just looking at a picture
like Figs. 4, since such plot can only be obtained,
if the location of the substates is known beforehand.
on that subspace optimally resolve the three substates. Note that it
would have not been possible to
identify number and location of the three substates
by just looking at a picture
like Figs. 4, since such plot can only be obtained,
if the location of the substates is known beforehand.
The substate picture permits a
drastically simplified description of protein dynamics, if the
dynamics within each substate can be considered irrelevant.
For our simplified protein model, the low frequency
conformational transitions can be described as a three state
Markov process (Fig.6), which has been shown
employing a statistical analysis of the nine
transition rates 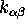 ,
,
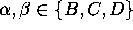 . These rates
were obtained from a
. These rates
were obtained from a 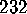 ns MD-simulation using the conformational
coordinates defined above[].
ns MD-simulation using the conformational
coordinates defined above[].
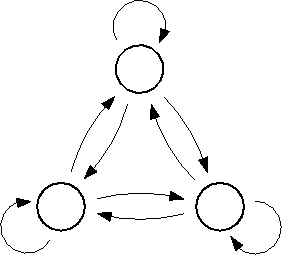
Figure 6: The low frequency conformational dynamics of the protein model
can be modeled as a Markov process with three states,
B,C and D, and nine transition rates, 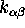 ,
,
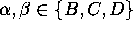 . A statistical analysis of 236
conformational transitions observed during a
. A statistical analysis of 236
conformational transitions observed during a 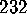 ns simulation
showed that the dynamics of the model,
if viewed on this coarse grained level, is indeed memory-free.
ns simulation
showed that the dynamics of the model,
if viewed on this coarse grained level, is indeed memory-free.
5. CONFORMATIONAL COORDINATES FROM PRINCIPAL COMPONENT ANALYSIS
We now turn our attention towards the conformational dynamics within
one conformational substate, which has been neglected in the above
Markov model. For our protein model,we chose
substate D (cf. Fig. 4). Accordingly,
we select only those configurational vectors out of the  ns
simulation for further analysis, which are closest to that substate.
ns
simulation for further analysis, which are closest to that substate.
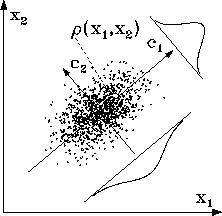
Figure: A two-dimensional example illustrates the method of
principal component analysis (PCA): For a given set of vectors (points),
which approximate a density  , PCA yields as eigenvectors
the direction of largest variance (
, PCA yields as eigenvectors
the direction of largest variance ( ), the direction of second
largest variance (
), the direction of second
largest variance ( ), and so on. The corresponding eigenvalues
(
), and so on. The corresponding eigenvalues
(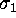 ,
,
 , ...)
measure the variances of the data set along these directions.
In the figure, these variances are indicated by the two Gaussians.
, ...)
measure the variances of the data set along these directions.
In the figure, these variances are indicated by the two Gaussians.
The structure of the free energy minima exhibited in (cf. Fig. 4) suggests that the corresponding density clusters can be described well by a --- possibly multivariate --- Gaussian distribution in configurational space. Under that assumption a principle component analysis (PCA)[15] can provide further information on the structure of that `cloud'. As a reminder, Fig. 7 illustrates the PCA-method using a two-dimensional data set.
Figure 8: A PCA-analysis was carried out on substate `D' of
the simplified protein model. The figure shows a selection of six out of
a total of 300 projections of the configuration space density along the
eigenvectors, ordered in decreasing variance;
 (a) (largest variance),
(a) (largest variance),
 (b),
(b),
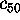 (c),
(c),
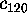 (d),
(d),
 (e), and
(e), and
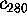 (f), respectively. A Gaussian
fit is shown with dashed lines for each of the projections. Note that
the scale of the abscissa in pictures (d-f) differs from that
in pictures (a-c).
(f), respectively. A Gaussian
fit is shown with dashed lines for each of the projections. Note that
the scale of the abscissa in pictures (d-f) differs from that
in pictures (a-c).
The results of a PCA of substate D are shown below. In the following,
the eigenvalues,
 , and the corresponding eigenvectors,
, and the corresponding eigenvectors,
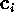 ,
shall be ordered in decreasing size.
Fig. 8 shows a sample of six projections of
,
shall be ordered in decreasing size.
Fig. 8 shows a sample of six projections of 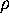 along
the eigenvectors
along
the eigenvectors 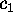 (largest variance),
(largest variance),
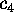 ,
,
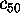 ,
,
 ,
,
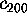 , and
, and
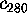 (smallest variance), respectively.
As can be seen from the fits (dashed), all projections are nearly Gaussian,
which assures, that PCA can be reasonably applied.
(smallest variance), respectively.
As can be seen from the fits (dashed), all projections are nearly Gaussian,
which assures, that PCA can be reasonably applied.
Fig. 9 shows a histogram of the eigenvalues 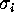 ,
,
 obtained from the PCA-analysis. These
eigenvalues measure the variance of substate-`cloud' along those
directions in configurational space, which are given by the corresponding
eigenvectors.
obtained from the PCA-analysis. These
eigenvalues measure the variance of substate-`cloud' along those
directions in configurational space, which are given by the corresponding
eigenvectors.
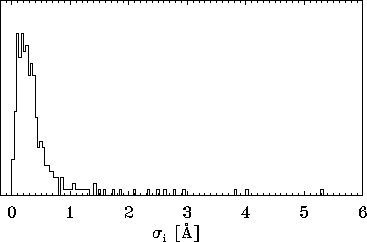
Figure: A histogram of standard deviations 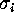 , derived from
the 300 eigenvalues given by the PCA analysis described in the text
reveals, that the vast majority of the
, derived from
the 300 eigenvalues given by the PCA analysis described in the text
reveals, that the vast majority of the 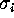 is smaller than
is smaller than
 Å , i.e., the configuration space density is `flat'
in most directions of configurational space. Only few extensions are
larger than
Å , i.e., the configuration space density is `flat'
in most directions of configurational space. Only few extensions are
larger than 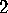 Å and contribute to the large-amplitude
motions observed in the protein model.
Å and contribute to the large-amplitude
motions observed in the protein model.
The surprising observation is that the cluster exhibits
large extensions (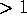 Å ) only along very few directions, whereas
it is `flat' (
Å ) only along very few directions, whereas
it is `flat' (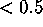 Å ) in 90% of the directions. Obviously,
only few degrees of freedom (namely those given by the `large'
eigenvectors) contribute significantly to the large amplitude
conformational dynamics of the protein model. Can these few
degrees of freedom be considered as relevant degrees of freedom
for the conformational dynamics? If so, can the remaining
90% degrees of freedom be considered as `noise', which need not
be included explicitly in a protein dynamics description?
Å ) in 90% of the directions. Obviously,
only few degrees of freedom (namely those given by the `large'
eigenvectors) contribute significantly to the large amplitude
conformational dynamics of the protein model. Can these few
degrees of freedom be considered as relevant degrees of freedom
for the conformational dynamics? If so, can the remaining
90% degrees of freedom be considered as `noise', which need not
be included explicitly in a protein dynamics description?
We cannot answer that questions yet; work is in progress.
It is clear, however, that the PCA serves to separate time scales, a
prerequisite for a dimension-reduced, effective description.
As an example, Fig. 10 compares part of our MD-simulation
projected on a `large' eigenvalue, 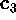 , (upper left
picture) with a projection on a `flat' eigenvalue,
, (upper left
picture) with a projection on a `flat' eigenvalue, 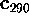 ,
(upper right). The bottom row of Fig. 10 shows
blow ups of the small rectangles in the upper two pictures.
,
(upper right). The bottom row of Fig. 10 shows
blow ups of the small rectangles in the upper two pictures.
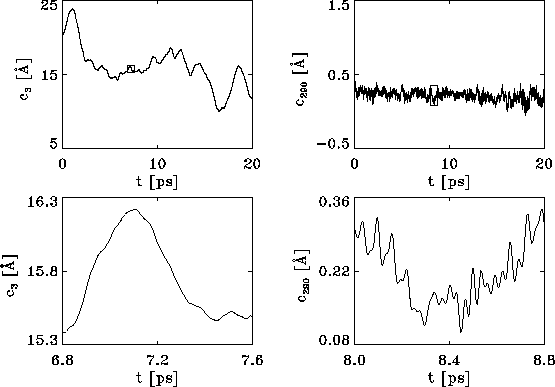
Figure 10: Upper row: Plots of the protein dynamics, projected
onto eigenvectors  (left side) and
(left side) and
 (right), respectively,
reveal that slow motions occur predominantly along directions in
configurational space characterized by large variances, whereas
fast motions are described by directions of small variance.
Lower row: Blow-ups of part of the dynamics (marked by small
rectangles in the upper two pictures) demonstrate that virtually
no high-frequency contributions of the molecular dynamics
are present in the large variance collective motions.
(right), respectively,
reveal that slow motions occur predominantly along directions in
configurational space characterized by large variances, whereas
fast motions are described by directions of small variance.
Lower row: Blow-ups of part of the dynamics (marked by small
rectangles in the upper two pictures) demonstrate that virtually
no high-frequency contributions of the molecular dynamics
are present in the large variance collective motions.
As can be seen, the high frequency parts of the dynamics are only present in the `flat' degrees of freedom, whereas the low frequency parts are captured by the `large' eigenvalues. Vice versa, no high frequency fluctuations are visible in the `large' eigenvalue dynamics, and no low frequency fluctuation occurs in the `flat' directions.
In order to quantify this observation, Fig. 11 shows
Fourier spectra of five projections on the eigenvectors
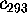 ,
,
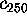 ,
,
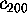 ,
,
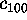 , and
, and
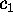 , respectively.
Again, the predominant contributions to the atomic fluctuations
(hatched areas) occur in the high frequency part in the case of the
`flat' degrees of freedom, whereas the dynamics along `large' eigenvalues
is exhibits merely low frequency contributions.
, respectively.
Again, the predominant contributions to the atomic fluctuations
(hatched areas) occur in the high frequency part in the case of the
`flat' degrees of freedom, whereas the dynamics along `large' eigenvalues
is exhibits merely low frequency contributions.
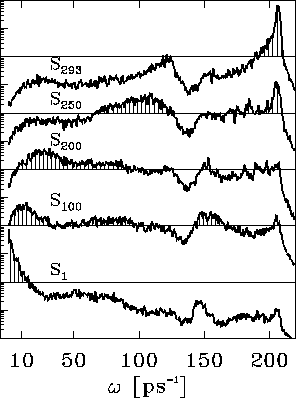
Figure 11: Predominant contributions to atomic fluctuations
(hatched areas) are
shifted from high frequencies to low frequencies as one
proceeds from small variances in configurational space
to large ones. The figure shows five
Fourier spectra of the protein dynamics, which has been projected
onto the eigenvectors (from top to bottom) 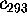 ,
,
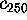 ,
,
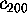 ,
,
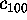 , and
, and
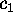 , respectively.
, respectively.
6. SUMMARY AND CONCLUSION
We illustrated concepts which aim at a reduction of the computational
effort, that has to be spent in the attempt to understand protein function
on the basis of protein dynamics. We illustrated these concepts using
extended molecular dynamics simulations of a simplified protein model.
Based on a thermodynamic formulation of the concept of conformational
substates in terms of minima in free energy, two methods to define
conformational coordinates, which serve to separate relevant from
irrelevant degrees of freedom, were explained and applied to a simplified
protein model.
The first method, the application of a neural clustering algorithm, allowed to identify substate hierarchies and led, in the case of the simplified protein model, to a Markov model for conformational transitions.
The second method served to analyze the dynamics within a conformational
state. It was shown that only few degrees of freedom contribute to
the large fluctuations in the protein model. At the same time,
these large fluctuations embody the low frequency contributions
to the protein dynamics, whereas all other degrees of freedom
carry the high frequency aspects. These findings should enable
the construction of effective descriptions for protein dynamics,
which permit longer simulation times and thereby extend the
range of biochemical processes that can be studied theoretically.
Acknowledgements
This work was supported
by the Deutsche Forschungsgemeinschaft (SFB 143/C1).